

Digital escape room
February 27, 2021
Last year was a bizarre year and, hopefully, one of a kind. Not only did Covid-19 affect our personal and professional lives, it also had a huge impact on regulatory inspections which were delayed or executed remotely.
In this article I present the main result of a data analysis on pharmaceutical FDA warning letters of 2020. I used the same tools as presented in one of my earlier articles on FDA warning letters.
As shown on the pie chart below 21CFR 211 (75 times) and 21CFR 201(73 times) are most cited in 2020 warning letters. Other parts which are regularly cited include 21CFR 330 (15 times) and those for medical devices 21CFR 820 (11 times), 21CFR 803 (7 times) and 21CFR 807 (5 times).
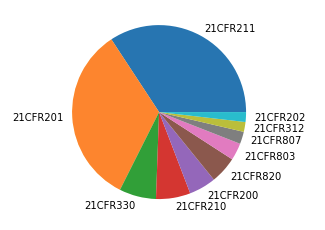
In the next sections I will look into the most cited paragraphs and the most common observations for parts 21CFR 211, 21CFR 201 and 21CFR 800.
21CFR 211 most cited paragraphs and observations
In 21CFR 211 “Current good manufacturing practice for finished pharmaceuticals” the most cited paragraphs of 2020 are shown in the bar-chart below.
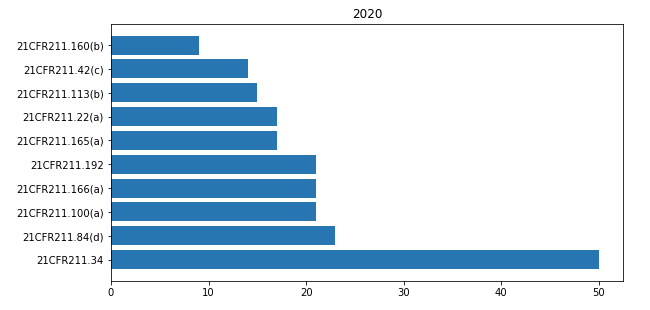
Paragraph 21CFR 211.34 is most cited. But in none of the warning letters this concerns an actual observation. From 2017 on many FDA warning letters include this as a general statement: “Based upon the nature of the violations we identified at your firm, we strongly recommend engaging a consultant qualified as set forth in 21CFR 211.34, to assist your firm in meeting CGMP requirements.”
Excluding 21CFR 211.34 let’s have a look at the most common observations that I found:
Your firm failed to validate and establish the reliability of your component supplier’s test analyses at appropriate intervals (21CFR 211.84(d)(1) and (2)).
Your firm failed to establish adequate written procedures for production and process control designed to assure that the drug products you manufacture have the identity, strength, quality, and purity they purport or are represented to possess (21CFR211.100(a)).
Your firm failed to establish a written testing program designed to assess the stability characteristics of drug products and to use results of such stability testing to determine appropriate storage conditions and expiration dates (21CFR211.166(a)).
Your firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed ( 21CFR211.192).
21CFR 201 most cited paragraphs and observations
In 21CFR 201 “Labeling” the most cited paragraphs of 2020 are shown in the bar-chart below.
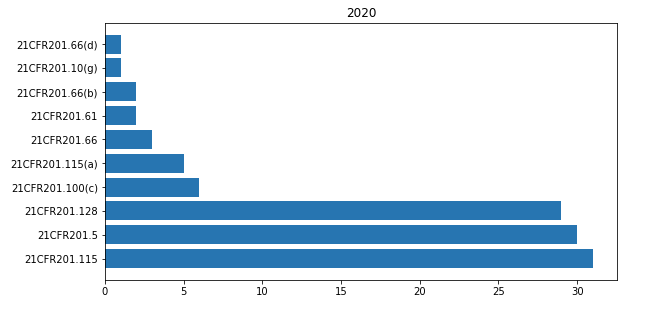
The most common observations that I found in FDA warning letters relating to this part are shown here below.
Your ineligible drug products are not exempted from the requirements of section 502 (f)(1) of the FDCA by regulations issued by the FDA (see , e.g., 21CFR201.115).
In addition, because these drugs are not approved in the U.S., they are also not exempt under 21CFR201.115 from the requirements of section 502(f)(1) of the FD&C Act .
Examples of claims observed on the BERSIH Antiseptic Alcohol 70 % Topical Solution Hand Sanitizer product label , that provide evidence of the intended use (as defined in 21CFR201.128 ) of the product include, but may not be limited to , the following : “ Hand Sanitizer “.
21CFR 800 most cited paragraphs and observations
In 21CFR 800 “Medical devices” the most cited paragraphs of 2020 are shown in the bar-chart below.
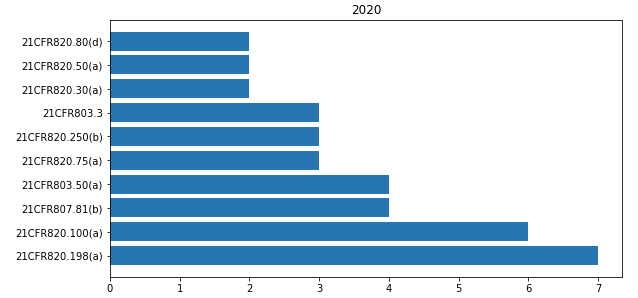
The most common observations that I found relating to this part are shown here below.
Failure to adequately establish procedures for receiving , reviewing , and evaluating complaints by a formally designated unit as per 21CFR820.198(a).
Failure to establish and maintain procedures for implementing corrective and preventive actions , as required by 21CFR820.100(a).
Failure to submit a report to the FDA no later than 30 calendar days after the day that your firm received or otherwise became aware of information, from any source , that reasonably suggests that a device that it markets has malfunctioned and this device or a similar device that your firm markets would be likely to cause or contribute to a death or serious injury, if the malfunction were to recur , as required by 21CFR803.50 (a)(2).
If you need further insights or similar data, don’t hesitate to get in touch with me or one of my colleagues at Trevalco.


{kind=link}
{kind=link}
{kind=link}